
Aperçu de la métagénomique 16S
Métagénomique
La métagénomique 16S est un domaine d'étude qui vise à comprendre la diversité des micro-organismes dans un échantillon donné en utilisant la technologie de séquençage à haut débit. Le principe de cette approche est que le gène de l'ARN ribosomal (ARNr) 16S, présent dans toutes les bactéries, peut être amplifié et séquencé pour identifier et quantifier les bactéries présentes dans un échantillon. La technique de la métagénomique 16S comprend les étapes suivantes :
- Collecte et conservation de l'échantillon
- Extraction de l'ADN de l'échantillon
- Isoler et purifier les protéines bactériennes d'intérêt prédites.
- Amplification par PCR du gène de l'ARNr 16S
- Séquençage de l'ADN amplifié
- Analyse et interprétation des données
L'analyse des données en métagénomique 16S implique le traitement des données séquencées à l'aide d'outils et d'algorithmes bioinformatiques. Les données traitées sont ensuite comparées à des bases de données de référence pour identifier les espèces bactériennes présentes dans l'échantillon et quantifier leur abondance relative.

Applications de la métagénomique 16S
- La compréhension de la diversité microbienne dans différents environnements, tels que le sol, l'eau et l'intestin humain.
- L'étude des effets des changements environnementaux sur les communautés microbiennes.
- L'identification de biomarqueurs pour diverses maladies et troubles.
- Le développement de nouvelles applications biotechnologiques, telles que les biocarburants et la biorémédiation.
Les données traitées obtenues à partir du séquençage métagénomique 16S sont analysées à l'aide de divers outils et algorithmes bioinformatiques, tels que le contrôle de la qualité, le filtrage, le regroupement et la classification taxonomique.
Les données dont la qualité a été contrôlée sont ensuite comparées à des bases de données de référence, telles que le Ribosomal Database Project (RDP) ou la base de données Greengenes, afin d'identifier les espèces bactériennes présentes dans l'échantillon et de quantifier leur abondance relative. Ces informations peuvent ensuite être visualisées à l'aide de diverses représentations graphiques, telles que des cartes de chaleur, des diagrammes à barres et des cladogrammes.
Les résultats de l'analyse métagénomique 16S peuvent fournir des informations importantes sur la composition et la diversité des communautés microbiennes dans un échantillon donné, ainsi que sur leurs rôles fonctionnels potentiels et leurs interactions au sein de la communauté.
Les résultats de l'analyse métagénomique 16S peuvent fournir des informations importantes sur la composition et la diversité des communautés microbiennes dans un échantillon donné, ainsi que sur leurs rôles fonctionnels potentiels et leurs interactions au sein de la communauté.
Outils pour l'analyse métagenomique
Il existe plusieurs outils et plateformes bioinformatiques couramment utilisés pour l'analyse des données de séquençage métagénomique 16S, notamment :
- QIIME (Quantitative Insights into Microbial Ecology) - Un logiciel open-source qui fournit un pipeline complet pour l'analyse des données métagénomiques 16S, du contrôle de la qualité à la classification taxonomique et à l'analyse fonctionnelle.
- DADA2 Un outil bioinformatique pour l'analyse des données de séquençage d'amplicons, y compris le séquençage du gène de l'ARNr 16S. DADA2 fournit des méthodes précises et efficaces pour le filtrage, le regroupement et la classification taxonomique des séquences d'amplicons.
- UPARSE Un algorithme rapide et précis pour le regroupement et la classification taxonomique des séquences d'amplicons du gène de l'ARNr 16S. UPARSE est un outil largement utilisé pour le traitement des données métagénomiques 16S à haut débit.
- Mothur Un logiciel libre qui fournit un pipeline complet pour le traitement, l'analyse et la visualisation des données métagénomiques 16S, y compris le contrôle de la qualité, le regroupement, la classification taxonomique et l'analyse des communautés.
- Metaphlan Un outil logiciel qui utilise une approche basée sur les k-mer pour la classification taxonomique des données métagénomiques 16S. Il est conçu pour traiter des ensembles de données métagénomiques importants et complexes et fournit une classification taxonomique au niveau des espèces et des souches.
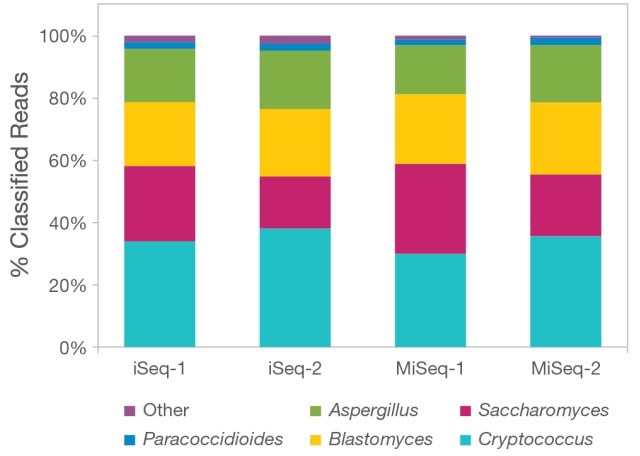
Ces outils sont parmi les plus couramment utilisés pour l'analyse des données métagénomiques 16S, mais il en existe de nombreux autres. Le choix de l'outil dépendra des exigences spécifiques de l'analyse, telles que la taille et la complexité de l'ensemble de données, ainsi que le niveau de précision et de détail souhaité dans les résultats.
{kind=link}
0 Commentaires